tl;dr AlphaFold2, an AI program developed by DeepMind, completely blew away the competition in a protein-structure prediction competition, CASP, so much so, the co-founder of the competition has commented that “in some sense” the 50-year old problem, is solved.
1. What is protein folding?
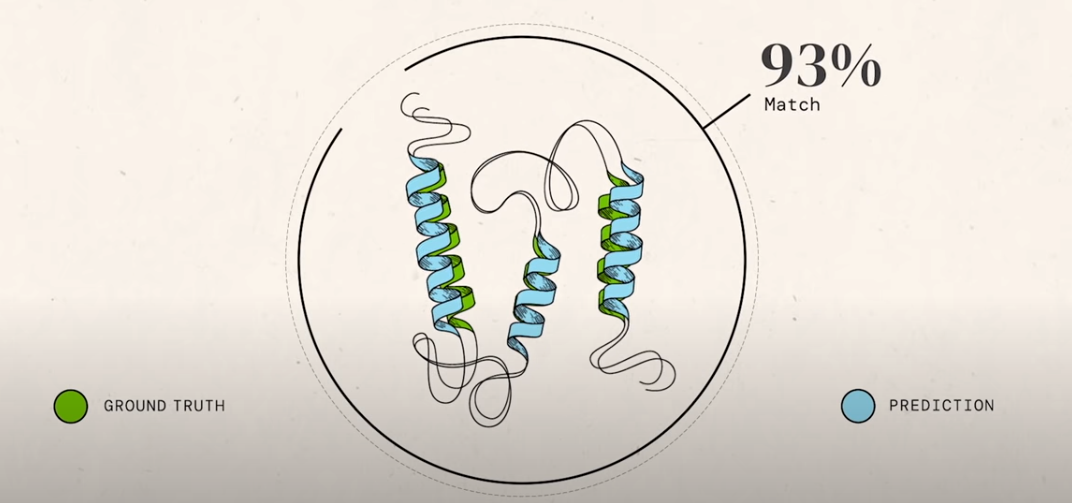
A protein’s function is determined by its structure, its 3D shape. The process, protein folding, is how a protein chain acquires this 3D shape and structure. The chain can easily contain hundreds of amino acids.
2. What is CASP and what were the results?
CASP (Critical Assessment of Structure Prediction) is a biennial blind protein-structure prediction competition where computational biologists try to predict the structure of several proteins whose structure has already been determined experimentally but not yet publicly released. This year marked the 14th edition of the competition.

AlphaFold2 was the huge winner of the competition, beating over 100 other teams. The above figure shows the sum of Z-scores representing the different participating groups. The Z-score is the difference of a sample’s value with respect to the population mean, divided by the standard deviation. A high value represents a large deviation from the mean. Groups with larger Z-scores, like AlphaFold2 (Group 427), whose average Z-score was around 2.5 when considering all targets, and rose to 3.8 on the hardest ones, performed much better than the average.
The actual performance (not relative to the other teams) is quite impressive too and has already been covered in great detail elsewhere and debated upon widely based on the little we know without the actual code and paper released. More on this when we tackle the debate on whether the problem is really solved.
AlphaFold2 as its name suggests is the second iteration of DeepMind’s AlphaFold, which debuted in the previous iteration of CASP in 2018. It is a huge improvement on that model, which was already state-of-the-art. Again, we don’t know the exact details till the paper is out, but more on this when we talk about the algorithm below.
A "gargantuan" leap today for #AI life science. @DeepMind @GoogleAI #AlphaFold2 prediction of #3D protein structure from amino acidshttps://t.co/UGlQByPu6k by @ewencallaway @NatureNews https://t.co/KxTVt8IKNa @demishassabis pic.twitter.com/RAoA9trsad
— Eric Topol (@EricTopol) November 30, 2020
3. What does this have to do with ImageNet?
Biology's ImageNet moment has arrived: #AlphaFold2 solves Protein Folding at the #CASP14 competition!
— Oriol Vinyals (@OriolVinyalsML) November 30, 2020
Blog: https://t.co/KH9g8FFXvS pic.twitter.com/IrNyZmva9V
DeepMind research scientist Oriol Vinyals termed the breakthrough as “Biology’s ImageNet moment”. The AI/ML community often recognises the work of the image dataset, ImageNet, and the ImageNet Large Scale Visual Recognition Challenge (ILSVRC) as setting the stage for landmark computer vision and ML achievements such as AlexNet in 2012 (which was 41% better than the next best competitor) and 2015’s ResNet and the “Deep Learning” era in ML research. NLP also had its ImageNet moment just a while back.
4. What are the implications of this breakthrough?
In short, this speeds up scientists work in understanding diseases, finding treatments, determining which drugs might be applied. On the other hand, some have brought up the possibility of its misuse, whether such a tool might be somehow weaponized.
In terms of drug discovery, drugs work by attaching a protein in a particular place, thereby altering or disabling its function. Knowing a protein’s shape may allow scientists in the future to identify such binding sites and make it easier to synthesise new and effective therapeutics. There is some debate though, on whether this is actually a rate-limiting factor in current drug discovery pipelines. What we do know is the anecdote from Andrei Lupas, an evolutionary biologist at the Max Planck Institute.
He had spent a decade trying to figure out the shape of one protein in a tiny bacteria-like organism called an archaeon, with AlphaFold2, he found the answer in half an hour. Wow!
5. Is the problem really solved?
Stephen Curry, a Professor of Structural Biology at Imperial College London seems to think otherwise and posted an article on his blog that acknowledges that Alphafold2 “will certainly help to advance biology” but “despite some of the claims being made, we are not at the point where this AI tool can be used for drug discovery.”
“For delivering reliable insights into protein chemistry or drug design” the root-mean-squared difference (RMSD) in atomic positions between the prediction and the actual structure needs to go down from 1.6 Å (0.16 nm) to within a margin of around 0.3Å.
Some practitioners seem to think otherwise though, with some accusations of “goal-post shifting”.
“As a practitioner, what I want to be able to do is pull a sequence that I’ve retrieved from DNA, drop it into a computer, and get the structure out. Intuitive insight can FOLLOW from those results.”
We will also expect DeepMind to be working out “corner cases” and improving the model as they decide how they want to use or release the model.
These are for single domains-not whole proteins-and there are a few poor predictions. So corner cases remain but core problem appears solved: 88% of predictions are <4Å, 76% <3Å, 46% <2Å. Unlike last time where there was some competition, this time AF2 was best for 88/97 targets.
— Mohammed AlQuraishi (@MoAlQuraishi) November 30, 2020
6. What was the algorithm used? How was the model trained?
Until the peer-reviewed paper is released and (if) the code is released, we don’t know much yet. As always, Yannic Kilcher does a great job of explaining the original AlphaFold paper in the video below.
Architecture-wise, for AlphaFold2, we don’t know. We do know that AlphaFold2 uses some sort of attention mechanism, maybe transformers replacing the ConvNet. It also seems they now fit over not just the fragements but the whole shape of the protein.
As mentioned in the DeepMind blog post they trained this system on ~170,000 protein structures from the protein data bank together with large databases containing protein sequences of unknown structure. The experimental setup for training was 16 x TPUs (roughly equivalent to 100-200 GPUs) run over a few weeks.
7. Will there be source code?
Until DeepMind decides how or if it wants to release whole or part of its code, we can only wait for the paper to be released.
DeepMind did release part of the original AlphaFold code (although in practice, researchers have pointed out “good luck generating the input features for something other than the CASP13 proteins”) and there have been open source implementations of the proposed model. MiniFold and AlphaFold Pytorch are some notable efforts.
8. How big was DeepMind’s funding and team and how does this compare to the Big Pharmas?
People have put the size of DeepMind at around 1000 people and perhaps 15-30 or so research teams. The video published by DeepMind showed 20 or so researchers on this particular team. The team was said to be cross-disciplinary and also includes researchers who have done PhDs or postdocs in Biology-related fields.
Big Pharma companies are said to spend at least hundreds of millions on R&D. They don’t usually get to hire the best minds in AI/ML though for much more specific, targeted research. It does make sense that this breakthrough comes from a pretty well-funded AI research laboratory rather than Big Pharma or academia.
9. What does this mean for AI/ML?
As reported in the tech media, it seems apparent that DeepMind has finally gone from “playing video games to addressing scientific problems with real-world significance — problems that can be life-or-death”.
The buzz that this breakthrough has created around the scientific and AI/ML communities is also just wonderful. It’s nice to end a quite terrible 2020 on a high note and it’s nice to see an AI/ML lab committed to deep research on difficult problems finding more success.
10. What is the link to the ML community in Singapore?
DeepMind co-founder + CEO, game developer and chess champion Demis Hassabis has some Singaporean heritage.
In a recent interview with the BBC, Demis, mentions that he took some early inspiration from, Foldit, a crowdsourcing puzzle game. Gamers who played it were actually trying to turn the protein into a particular shape, which led to the discovery of a few important structures for real proteins. So also a shout out to gamers in the community!